cat P06780.fasta16 Predicción de estructuras
En este módulo aprenderás a interpretar los valores de confianza y precisión de los modelos.
16.1 🧾 Entrada en formato FASTA
Toda predicción estructural parte de una secuencia en formato FASTA, por ejemplo:
⚠️ Asegúrate de que:
- La secuencia esté limpia (sin caracteres inválidos).
- Tenga un número suficiente de residuos (recomendado: > 50 aa).
- El encabezado comience con
>seguido de un identificador
16.2 📈 Interpretación de resultados
Las herramientas de predicción devuelven archivos con extensiones como .pdb, junto con métricas de calidad del modelo.
16.2.1 🟨 pLDDT (per-residue Local Distance Difference Test)
- Significado: predice qué tan confiable es la posición de cada átomo respecto al modelo.
- Visualización: colores aplicados directamente sobre el modelo estructural (azul = alta confianza, rojo = baja).
- Escala: 0–100.
| pLDDT | Confianza |
|---|---|
| >90 | Muy alta |
| 70–90 | Alta |
| 50–70 | Baja |
| <50 | Muy baja (desorden) |
🧠 En proteínas, se aproxima a lDDT-Cα pero con mayor precisión. Para ácidos nucleicos, se usa un radio de 30 Å. Para ligandos, solo se consideran distancias con los polímeros, no otros ligandos.
🌟 Puedes visualizar el modelo con colores según pLDDT en ChimeraX o PyMOL para detectar regiones confiables o móviles.
16.2.2 🧩 PAE (Predicted Aligned Error)
- Significado: estimación del error esperado entre dos regiones (i, j) del modelo.
- Formato: matriz cuadrada simétrica que representa la incertidumbre en la relación entre pares de residuos.
- Unidades: Ångstroms (distancia).
- Útil para evaluar relaciones entre dominios o subunidades.
- Se interpreta como una mapa de calor, donde los valores bajos indican mayor confianza estructural.
🔬 Muy útil para saber si dos regiones están bien orientadas entre sí (por ejemplo, dominios móviles, loops flexibles, etc.).
Visualizable como mapa de calor:
| Valor PAE | Confianza relacional |
|---|---|
| <5 Å | Alta |
| >10 Å | Baja |
16.2.3 🧬 pTM e ipTM – Evaluación del modelo global
- pTM (predicted TM-score):
- Mide la precisión global del modelo.
- Útil para evaluar si el plegamiento total es confiable.
- Valores: 0 a 1.
- 0.5 = el modelo es similar a la estructura real.
- <0.5 = baja precisión global.
- Valores: 0 a 1.
- ipTM (interfacial predicted TM-score):
- Evalúa la precisión de la orientación relativa entre subunidades (en complejos).
- Ideal para estructuras multímero o ensamblajes.
| Score | Interpretación |
|---|---|
| >0.8 | Alta confianza estructural |
| 0.6–0.8 | Zona gris, posible confianza |
| <0.6 | Predicción probablemente fallida |
🔬 Nota: con secuencias muy cortas (<20 residuos), pTM puede no ser confiable. En ese caso, se prioriza el uso de pLDDT y PAE.
16.3 Ejemplo
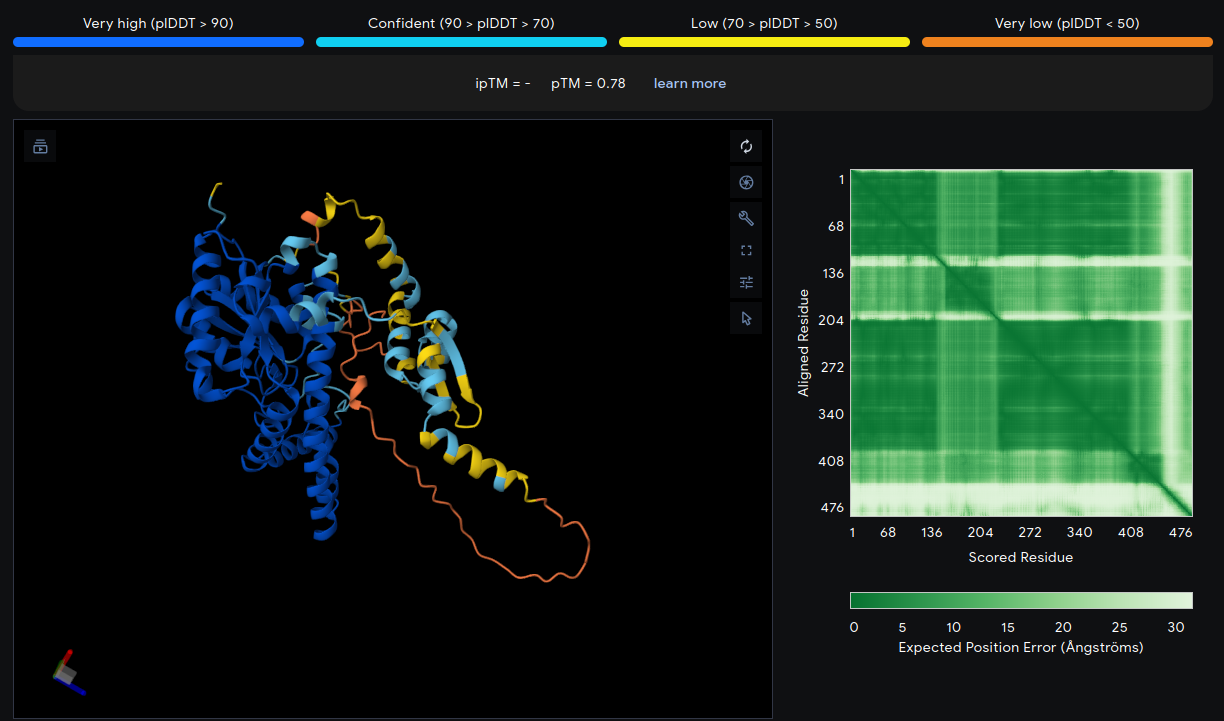
16.4 📦 Archivos de salida típicos
| Archivo | Contenido |
|---|---|
model.pdb |
Modelo estructural en formato 3D estándar |
ranking_debug.json |
Ranking de confianza de los modelos generados |
plddt_scores.txt |
Confianza por átomo o residuo |
pae.png o pae.json |
Matriz visual o numérica del error entre residuos |
predicted_aligned_error |
Archivo numérico completo para análisis con scripts |
16.5 🧭 Recomendaciones
- Usa pLDDT para identificar regiones confiables y poco confiables.
- Analiza PAE si tu proteína tiene dominios móviles o interacción con otras.
- Usa pTM/ipTM para evaluar modelos completos o complejos multímero.
- Visualiza con ChimeraX para examinar colores por confianza.
16.6 📌 Conclusión
Las predicciones estructurales actuales permiten obtener modelos de alta calidad. Sin embargo, su interpretación debe combinar métricas locales (pLDDT), relacionales (PAE) y globales (pTM, ipTM) para asegurar un uso biológicamente informado y confiable del modelo.